
Arteriosklerose
Seite 5 von 10
| <<< zurück | >>> weiter |
ÖAK Fortbildungsdiplom
Cholesterin wird beim gesunden Menschen etwa zu einem Drittel mit der Nahrung aufgenommen und zu zwei Dritteln im Körper produziert. Es wird u.a. bei der Synthese von Sexualhormonen und Gallensäuren benötigt. Cholesterin und andere Blutfette wie z.B. Triglyzeride werden im Blut an Transporteiweiße, den sog. Lipoproteinen gebunden. Je nach Zusammensetzung und Dichte der Lipoproteine werden VLDL (Very Low Density Lipoproteine), LDL (Low Density Lipoproteine) und HDL (High Density Lipoproteine) unterschieden.
Aus derzeitiger Sicht ist das in den LDL gebundene Cholesterin an der Entstehung der Arteriosklerose maßgebend beteiligt. Es wird deshalb umgangsprachlich als das schlechte LDL-Cholesterin bezeichnet, da es nach der Synthese in der Leber in andere Organe und Gewebe gelangt und sich dort u.a. in den Gefäßen ablagert. Dagegen transportiert das gute HDL das Cholesterin zur Leber zurück. Daraus kann abgeleitet werden, dass eine niedrige Konzentration von LDL-Cholesterin (unter 4,0 mmol/l) im Blut erwünscht ist, während das HDL-Cholesterin (>0,9 mmol/l) möglichst hoch sein sollte. Eine Erhöhung des Gesamtcholesterin im Blut wird als Hypercholesterinämie bezeichnet. Sind nur die Triglyzeridwerte erhöht spricht man von einer Hypertriglyzeridämie.
Fast alle Köperzellen wie z.B. Muskel- und Fettgewebe sind durch einen speziellen Rezeptor in der Lage, das LDL-Cholesterin aus dem Blut aufzunehmen. In der Zelle wird das Cholesterin u.a. zur Hormonsynthese verwendet. Überschüssiges Cholesterin wird gespeichert oder von den HDL aufgenommen und wieder abtransportiert.
Auf zellulärer Ebene werden bei der Entwicklung der Arteriosklerose derzeit folgende Mechanismen postuliert (Lipidtherorie).
- Voraussetzung
für die Entstehung eines arteriosklerotischen Prozesses (Läsion)
ist ein Endothelzellschaden der Arterienintima. An diesen Schadstellen
kommt es zu einer Anhaftung von Monozyten. Die Monozyten gehören
zu den weißen Blutzellen, also den Leukozyten, und haben die Eigenschaft,
nach kurzer Zirkulation im Blut in verschiedene Organe auszuwandern
und dort zu ortsständigen Makrophagen (umgangssprachlich Fresszellen)
zu differenzieren. Das Anhaften der Monozyten an den Endothelzellen
der Intima wird durch Adhäsionsmoleküle bewirkt.
- Durch
verschiedene Enzyme und Wachstumsfaktoren wird ein Eindringen der Monozyten
in die Intima und eine Umwandlung zu Makrophagen möglich. Makrophagen
sind wichtige Vermittler der Immunabwehr und können Mikroorganismen
und Zelltrümmer nach Aufnahme eliminieren (sog. Phagozytose). Wie
die meisten Zellen nehmen auch die in die Gefäßwand eingewanderten
Makrophagen das LDL-Cholesterin durch oberflächliche Rezeptoren
auf. Die eingewanderten Makrophagen wiederum produzieren weitere Wachstumsfaktoren
und Zytokine, also bestimmte Wachstumshormone, und bewirken somit einen
Einstrom von Lipiden, u.a. dem LDL-Cholesterin und weiterer Blutzellen.
Die Folge ist eine zunächst auf die Intima beschränkte Entzündungsreaktion,
die später auch auf die Muskelzellen der Media übergreift.
- Ein entscheidendes
Merkmal der Makrophagen wirkt sich dann bei einem erhöhten Angebot
von LDL-Cholesterin verhängnisvoll für das gesamte Gefäßsystem
aus: So besitzen die Makrophagen neben dem üblichen LDL-Rezeptor
(der einen Rückkopplungsmechanismus aufweist, wodurch die Cholesterin-Aufnahme
wieder gestoppt werden kann) einen zweiten LDL-Rezeptortyp. Dieser so
genannte Scavanger-Rezeptor (Scavenger [engl.] = Straßenfeger)
nimmt weiter ungebremst Cholesterin auf, wodurch sich die Makrophagen
in fettangereicherte Schaumzellen umwandeln, dem entscheidenden Bestandteil
der arteriosklerotischen Plaques. Dabei werden durch den Scavanger-Rezeptor
nur modifizierte LDL, wie z.B. oxidierte LDL aufgenommen.
- Das Aufbrechen
dieser arteriosklerotischen Plaques und die anschließende Auflagerung
eines Blutgerinnsels (Thrombus) ist die häufigste Ursache des akuten
Arterienverschlusses. Das Risiko für einen akuten Koronararterienverschluss
wird in erster Linie von der Plaquestabilität und weniger
vom Stenosegrad, also der Größe der Einengung, des Gefäßes
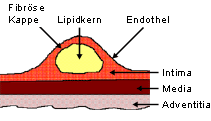
bestimmt. Besonders instabile Plaques weisen eine besonders dünne
bindegewebige Kappe auf, die einen großen Lipidkern bedeckt. Nach
einem Plaqueriss (Ruptur) kommt das Blut mit den hochgradig thrombogenen
Material des Lipidkerns in Kontakt, was die Entstehung des finalen Thrombos
bewirkt. Die Folge davon ist ein akuter Koronararterienverschluss mit
einem oft tödlich verlaufenden Myokardinfarkt.
 |
|
Schematischer Aufbau einer fortgeschrittenen arteriosklerotischen Plaques |
Diese Abbildung
zeigt den schematischen Aufbau einer fortgeschrittenen arteriosklerotischen
Plaque. Bei Einriss der fibrösen Kappe liegt der cholesterinhaltige
Lipidkern frei, wodurch sich in kurzer Zeit ein Thrombus auflagern kann.
Die Folge ist dann ein kompletter Gefäßverschluss.